SINCHON·INT′l
SINCHON·INT′l
- MIRAE

show mobile menu
mobile menu
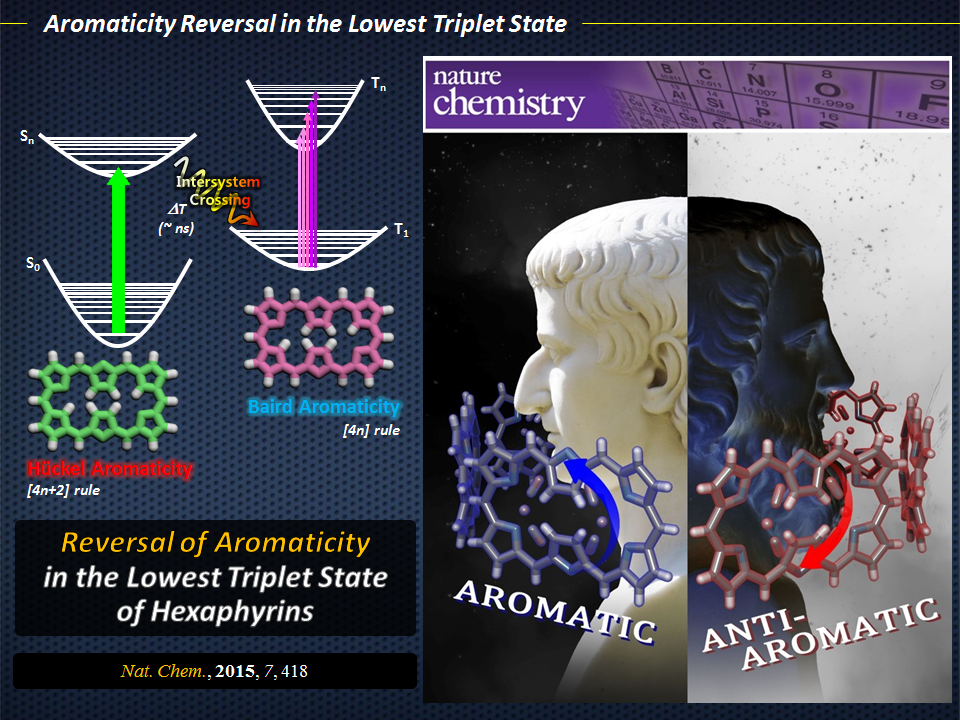
Aromaticity reversal in the lowest triplet state
A group of Yonsei researchers demonstrates that bis-rhodium hexaphyrins containing [26] and [28] π-electron peripheries show properties consistent with Baird’s rule
As any organic chemist will tell you, aromaticity is a fundamental concept because it relates to the chemical properties and reactivities of a wide range of species. At the most basic level, a compound (more specifically, a planar ring molecule) can be classified as aromatic, antiaromatic, or non-aromatic. The quantum mechanical basis for this classification was described by Hückel, whose [4n+2] rule defined a compound as being aromatic when its number of π-electrons is equal to 4n+2, where “n” is any positive integer. Since the introduction of this rule, various concepts of aromaticity have been reported, and a new rule for the lowest triplet state was proposed by Baird, where the Hückel (anti)aromaticity in the ground state would be reversed in the excited state. Despite the potential of excited-state aromaticity for application in areas such as photonic devices, there has been no report on the comparison between the electronic structures of the ground (closed-shell singlet) and excited (triplet excited) states of a pair of aromatic and antiaromatic congeners. Indeed, such studies would be useful to test Baird’s rule and experimentally confirm the reversal of (anti)aromaticity in the excited state.
Congeneric expanded porphyrins R26H and R28H
With the aforementioned background, the researchers analyzed a pair of congeneric expanded porphyrins (bis-rhodium [26]- and [28]hexaphyrins, R26H and R28H) with well-defined [4n+2] and [4n]π-electron systems in the ground state. These compounds are particularly useful as they can release or capture two pyrrolic protons following two-electron reduction or oxidation of the macrocycle, and hence can be employed to examine molecular aromaticity. Specifically, the photophysical properties of expanded porphyrins can be employed to determine aromaticity in [4n]/[4n+2]π-electron heteroannulenes. Generally, when compared to aromatic expanded porphyrins, the antiaromatic congeners exhibit broad, weak, and featureless absorption spectra. Moreover, they display a lack of fluorescence intensity and rapid decay from the excited state to the ground state. Indeed, as R26H and R28H represent Hückel aromatic and antiaromatic compounds, respectively, the intensities and spectral shapes of their transient absorption (TA) spectra differ greatly, and as such, they are considered suitable to explore the validity of Baird’s prediction. Moreover, the presence of coordinated bis-rhodium metals helps rigidify the nearly planar conformations of these chemically stable species, while the two coordinated rhodium metal cations should accelerate the intersystem crossing processes required to populate the corresponding excited triplet states through the heavy atom effect. Meanwhile, disturbance of the excited state dynamics should be minimized because of the closed-shell configuration of the rhodium(I) centers.
Spectral data and calculations
In contrast to the intense bands observed in the ground-state absorption spectrum of R26H, R28H exhibited relatively broad and weak spectra. Furthermore, the singlet excited-state lifetimes of these compounds were shorter than those of metal-free hexaphyrins due to efficient intersystem crossing (Sn→Tn). The decay-associated spectra reflecting the excited-state absorption (ESA) and ground-state bleaching of the transient species corresponding to the S1 and T1 states were obtained, and the extinction coefficients were estimated to compare the absorbance differences between the two states. The obtained ESA spectra of the T1 states of the two hexaphyrins further indicated reversal of the (anti)aromaticity: the spectrum of R26H was comparable to the spectra observed for antiaromatic porphyrinoids, while the spectrum of R28H resembled the spectra of canonical aromatic porphyrinoids.
As expected, the calculated vertical energy transitions of R26H and R28H revealed that the transitions of the triplet state R26Hresembled those of the ground state R28H, and vice versa. Calculated nucleus-independent chemical shift (NICS) values for the T1states of R26H and R28H indicated that diatropic and paratropic ring currents were present in the ground and T1 states of R26H, respectively. The opposite was true for R28H; hence, the NICS values accurately reflected the magnitude of the global or macrocycle-derived aromaticity. This was supported by the ACID (anisotropy of the induced current density) plots, where the overall directions of the ring current densities induced by external magnetic fields changed from clockwise (aromatic) to anticlockwise (antiaromatic) in the ground and T1 states of R26H, respectively. Again, the opposite trend was observed for R28H. Finally, the HOMA (harmonic oscillator model of aromaticity) values for all available 26- and 28-π-electronic circuits of R26H and R28H further confirmed the proposed reversal of (anti)aromaticity in the T1 state.
Reversal of aromaticity
The authors concluded that the spectroscopic and calculated features of Hückel aromatic and antiaromatic hexaphyrins in their T1states demonstrate the reversal of Hückel (anti)aromaticity. These findings are consistent with previous theoretical analyses of simple annulenes, such as benzene. As such, the study proposes the possibility of manipulating (anti)aromaticity through the selection of a desired electronic state to provide novel tools for modulating the chemical features and reactivity of highly conjugated systems. This could facilitate the development of fast saturable absorbers, novel molecular magnets, and photoswitchable devices that operate in both visible and NIR regions.
Recommended Articles


Professor Byeong-Su Kim
New study demonstrates that “deformable” electronics are not a stretch

Professor Yeonjin Yi


